

Research Area(s)
- HIV restriction factors, DNA deaminases, Mutagenesis, Enzyme mechanisms
About
TEDx: "How our changing DNA keeps us alive”
Areas of Expertise
HIV restriction factors, DNA deaminases, mutagenesis, enzyme mechanisms
In the News
- USask researchers paving the way to better HIV treatments
- Melfort Native Named Department Head at University of Saskatchewan
- When the immune system proteins go rogue
- Just Give Mutants a Chance: Dr. Linda Chelico
- From MUCC to microbiology: Dr. Linda Chelico working on HIV, cancer research projects
- Chelico looking for answers at molecular level
- USask scientist boosting natural bodyguards against HIV
- HIV research: CTV Video
- Young Innovators: HIV virus fighter linked to cancer mutations
- Human Protein Fights HIV as Monomer and Dimer, Says Study
- Unexpected cause of mutation in cancer identified
- Q5 with Dr. Linda Chelico and her team
- U of S researchers hope to harness human DNA to fight HIV
- Evolution of the arms race
Research Interests
Our lab has two main projects that examine host restriction factors that restrict viral replication and the origins of mutations in cancer cells. These seemingly disparate topics are unified by the enzyme family involved in both processes, the APOBEC3 family of cytosine deaminases.
Retrotransposons and endogenous retroviruses have been genomic parasites in organisms throughout evolution and have contributed to both species evolution and disease. The APOBEC (Apolipoprotein B mRNA-editing enzyme-catalytic polypeptide) family of enzymes present in their earliest form acted as a defense to retroelements. Due to expansion of retroelements through evolution there was a corresponding expansion in the APOBEC family. The most recent expansion in placental mammals formed the APOBEC-like 3 (APOBEC3) family in response to ancient pathogenic retroviruses. Humans contain seven APOBEC3 (A3) enzymes (A3A, A3B, A3C, A3D, A3F, A3G, and A3H).
The A3 enzymes act as host restriction factors to inhibit retroelement, e.g., LINE-1, retrovirus, e.g., HIV-1, DNA virus, e.g., EBV, or RNA virus, e.g., coronavirus, replication through either nucleic acid binding ability or activity as single-stranded (ss) DNA and RNA cytosine deaminases that catalyze the formation of promutagenic uracils. Our lab studies from a biochemical and cellular perspective how A3 enzymes restrict the replication of the retrovirus HIV-1.
Restriction of the replication of HIV-1 by A3 enzymes occurs through the ssDNA cytosine deamination activity of A3 enzymes which results in hypermutated and inactivated viral genomes (Figure 1). HIV-1 can suppress A3 restriction factors by encoding the accessory protein Vif that hijacks the host ubiquitination system to induce polyubiquitination and proteasomal degradation of A3 enzymes.
Our lab studies:
(1) The biochemistry of A3 enzymes and how A3 enzymes interact with each other and cellular and viral proteins to restrict viral replication.
(2) The biochemical interface of HIV-1 Vif and A3 enzymes and the determinants of Vif-mediated degradation of A3 enzymes.
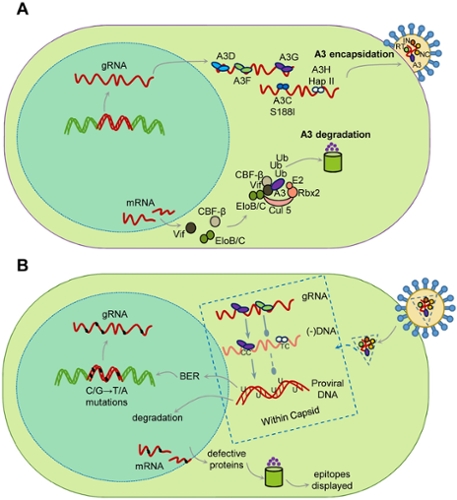

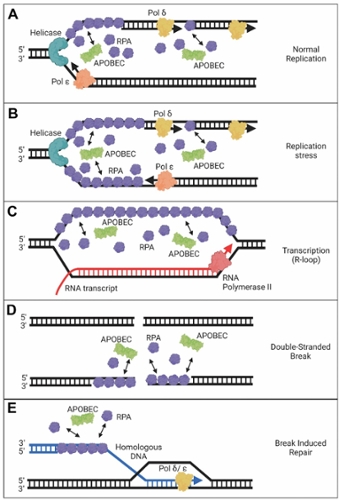
Despite these benefits of A3 enzymes for suppression of retroelements and viruses, there is evidence that there is a cost to this defense system in the form of A3-catalyzed deaminations that occur in our genomes during our lifetime. Usually, redundant DNA repair mechanisms can remove uracils and negate most of these promutagenic lesions. However, with the development of Next Generation Sequencing technology to obtain greater sequencing depth, it is clear that many cancer genomes have a bias of A3-induced mutations at cytosines and most cancer cells or tumors show overexpression of A3A, A3B, or A3H mRNA, suggesting that some uracils persist and develop into mutations.
Our lab studies:
(1) The mechanisms by which A3 enzymes gain access to transiently single-stranded DNA in the genome and compete with other single-stranded DNA binding proteins, such as replication protein A (RPA).
(2) The influence of A3-induced mutations on the fate of tumor cells and non-tumorigenic epithelial cells using mouse xenograft models.
(3) How interactions of A3 enzymes with other cellular proteins influence genomic stability and tumorigenesis.

Recent Publications
- Granadillo Rodríguez M, Wong L, Shayeganmehr A, Pellegrina D, Vizeacoumar FS, Vizeacoumar FJ, Helmy M, Chelico L. Differential effects on tumor progression by APOBEC3A, APOBEC3B, and APOBEC3H Haplotype I in a breast cancer mouse xenograft model. Frontiers in Genetetics. doi: 10.3389/fgene.2025.1425483. (2026)
- Pacheco J, Yousefi M, Yang H, Li S, Chelico L, Chen XS. Both Domains of APOBEC3F Recognize AA RNA Motifs to Support HIV-1 Virion Encapsidation and Antiviral Function. Journal of Molecular Biology. doi: 10.1016/j.jmb.2025.169536. (2025)
- Gaba A, Yousefi M, Bhattacharjee S, Chelico L. Variability in HIV-1 transmitted/founder virus susceptibility to combined APOBEC3F and APOBEC3G host restriction. Journal of Virology. doi: 10.1128/jvi.01606-24. (2024)
- Chelico L and Feng Y. In vitro deamination assay to measure the activity and processivity of AID/APOBEC enzymes. Methods in Enzymology. doi.org/10.1016/bs.mie.2024.11.038 (2024)
- Chelico L and Adolph MB. Purification of enzymatically active APOBEC proteins from an insect cell expression system. Doi.org/10.1016/bs.mie.2024.11.035 (2024)
- Jang GM, Annan Sudarsan AK, Shayeganmehr A, Prando Munhoz E, Lao R, Gaba A, Granadillo Rodríguez M, Love RP, Polacco BJ, Zhou Y, Krogan NJ, Kaake RM, Chelico L. Protein Interaction Map of APOBEC3 Enzyme Family Reveals Deamination-Independent Role in Cellular Function. Molecular and Cellular Proteomics. 23(5):100755. doi: 10.1016/j.mcpro.2024.100755 (2024).
- Bhattacharjee S, Gaba A, Chelico L. APOBEC3D excludes APOBEC3F from HIV-1 virions by competitive binding of RNA. Frontiers in Virology. doi.org/10.3389/fviro.2024.1343037 (2024).
- Granadillo Rodríguez M, Wong L, Chelico L. Similar deamination activities but different phenotypic outcomes induced by APOBEC3 enzymes in breast epithelial cells. Frontiers in Genome Editing. doi: 10.3389/fgeed.2023.1196697 (2023).
- Yousefi M, Annan Sudarsan AK, Gaba A, Chelico L. Stability of APOBEC3F in the Presence of the APOBEC3 Antagonist HIV-1 Vif Increases at the Expense of Co-Expressed APOBEC3H Haplotype I. Viruses. doi: 10.3390/v15020463 (2023).
- Liu W, Newhall KP, Khani F, Barlow L, Nguyen D, Gu L, Eng K, Bhinder B, Uppal M, Récapet C, Sboner A, Ross SR, Elemento O, Chelico L, Faltas BM. The Cytidine Deaminase APOBEC3G Contributes to Cancer Mutagenesis and Clonal Evolution in Bladder Cancer. Cancer Res. doi: 10.1158/0008-5472.CAN-22-2912 (2023)
- Wong L, Sami A, Chelico L. Competition for DNA binding between the genome protector replication protein A and the genome modifying APOBEC3 single-stranded DNA deaminases. Nucleic Acids Res. doi: 10.1093/nar/gkac1121 (2022)
- Barzak FM, Ryan TM, Mohammadzadeh N, Harjes S, Kvach MV, Kurup HM, Krause KL, Chelico L, Filichev VV, Harjes E, Jameson GB. Small-Angle X-ray Scattering (SAXS) Measurements of APOBEC3G Provide Structural Basis for Binding of Single-Stranded DNA and Processivity. Viruses. doi: 10.3390/v14091974 (2022).
- Gaba A, Hix MA, Suhail S, Flath B, Boysan B, Williams DR, Pelletier T, Emerman M, Morcos F, Andrés Cisneros G, Chelico L. Divergence in dimerization and activity of primate APOBEC3C. Journal of Molecular Biology. doi: 10.1016/j.jmb.2021.167306 (2021)
- Chelico L. Special Issue "APOBECs and Virus Restriction". Viruses. 13:1613 (2021).
- Kaake RM, Echeverria I, Kim SJ, Von Dollen J, Chesarino NM, Feng Y, Yu C, Ta H, Chelico L, Huang L, Gross J, Sali A, Krogan NJ. Characterization of an A3G-VifHIV-1-CRL5-CBFβ Structure Using a Cross-linking Mass Spectrometry Pipeline for Integrative Modeling of Host-Pathogen Complexes. Molecular and Cellular Proteomics. doi: 10.1016/j.mcpro.2021.100132 (2021).
- Gaba A, Flath B, Chelico L. Examination of the APOBEC3 Barrier to Cross Species Transmission of Primate Lentiviruses. Viruses. 13:1084 (2021).
- McDonnell MM, Karvonen SC, Gaba A, Flath B, Chelico L, Emerman M. Highly-potent, synthetic APOBEC3s restrict HIV-1 through deamination-independent mechanisms. PLoS Pathogens. 17:e1009523 (2021).
- Nchioua R, Kmiec D, Gaba A, Stürzel CM, Follack T, Patrick S, Kirmaier A, Johnson WE, Hahn BH, Chelico L, Kirchhoff F. APOBEC3F Constitutes a Barrier to Successful Cross-Species Transmission of Simian Immunodeficiency Virus SIVsmm to Humans. Journal of Virology. 95:e0080821 (2021).
- Wong L, Vizeacoumar FS, Vizeacoumar FJ, Chelico L. APOBEC1 cytosine deaminase activity on single-stranded DNA is suppressed by replication protein A. Nucleic Acids Res. 49:322-339 (2021).
- Granadillo Rodríguez M, Flath B, Chelico L. The interesting relationship between APOBEC3 deoxycytidine deaminases and cancer: a long road ahead. Open Biol. 10:200188 (2020).
- Conner KL, Shaik AN, Marshall KA, Floyd AM, Ekinci E, Lindquist J, Sawant A, Lei W, Adolph MB, Chelico L, Siriwardena SU, Bhagwat A, Kim S, Cote ML, Patrick SM. APOBEC3 enzymes mediate efficacy of cisplatin and are epistatic with base excision repair and mismatch repair in platinum response. NAR Cancer. 2:zcaa033 (2020).
- Hix MA, Wong L, Flath B, Chelico L, Cisneros GA. Single-nucleotide polymorphism of the DNA cytosine deaminase APOBEC3H haplotype I leads to enzyme destabilization and correlates with lung cancer. NAR Cancer. 2:zcaa023 (2020)
- Chelico L, King A, Ticknor J, McDonald M, Rosenes R, Mercredi J, Saddleback J, Bailey G, King M; Saskatoon Tribal Council Health & Family Services. Perspectives of Saskatchewan researchers and community members on HIV-1 strains circulating in Saskatchewan. AIDS. 34:1987-1989. (2020)
- Yang H, Ito F, Wolfe AD, Li S, Mohammadzadeh N, Love RP, Yan M, Zirkle B, Gaba A, Chelico L, Chen XS. Understanding the structural basis of HIV-1 restriction by the full length double-domain APOBEC3G. Nature Communications. 11:632 (2020)
- McDaniel YZ, Wang D, Love RP, Adolph MB, Mohammadzadeh N, Chelico L, Mansky LM. Deamination hotspots among APOBEC3 family members are defined by both target site sequence context and ssDNA secondary structure. Nucleic Acids Research. doi: 10.1093/nar/gkz1164. (2020)
- Morse M, Naufer MN, Feng Y, Chelico L, Rouzina I, Williams MC. HIV restriction factor APOBEC3G binds in multiple steps and conformations to search and deaminate single-stranded DNA. Elife. doi: 10.7554/eLife.52649 (2019)
- Mohammadzadeh N, Love RP, Gibson R, Arts EJ, Poon AFY, Chelico L. Role of co-expressed APOBEC3F and APOBEC3G in inducing HIV-1 drug resistance. Heliyon. 5:e01498 (2019)
- Adolph MB, Ara A, Chelico L. APOBEC3 Host Restriction Factors of HIV-1 Can Change the Template Switching Frequency of Reverse Transcriptase. Journal of Molecular Biology. 431:1339-1352 (2019)
- Mohammadzadeh N, Follack TB, Love RP, Stewart K, Sanche S, Chelico L. Polymorphisms of the cytidine deaminase APOBEC3F have different HIV-1 restriction efficiencies. Virology, 527:21-31. (2019)
- Feng Y, Wong L, Morse M, Rouzina I, Williams MC, Chelico L. RNA-Mediated Dimerization of the Human Deoxycytidine Deaminase APOBEC3H Influences Enzyme Activity and Interaction with Nucleic Acids. Journal of Molecular Biology, 430:4891-4907. (2018)
- Adolph MB, Love RP, Chelico L. Biochemical Basis of APOBEC3 Deoxycytidine Deaminase Activity on Diverse DNA Substrates. ACS Infectious Diseases 4:224-238. (2018)
- Morse M, Huo, R, Feng, Y, Rouzina I, Chelico L, Williams MC. Dimerization regulates both deaminase-dependent and deaminase-independent HIV-1 restriction by APOBEC3G. Nature Communications. doi:10.1038/s41467-017-00501-y (2017)
- Adolph MB, Love RP, Feng Y, Chelico L. Enzyme cycling contributes to efficient induction of genome mutagenesis by the cytidine deaminase APOBEC3B. Nucleic Acids Research. doi: 10.1093/nar/gkx832 (2017)
- Feng Y, Goubran MH, Follack TB, Chelico L. Deamination-independent restriction of LINE-1 retrotransposition by APOBEC3H. Scientific Reports. 7:10881. (2017)
- Adolph MB, Ara A, Feng Y, Wittkopp CJ, Emerman M, Fraser JS, Chelico L. Cytidine deaminase efficiency of the lentiviral viral restriction factor APOBEC3C correlates with dimerization. Nucleic Acids Research, Feb 1. doi: 10.1093/nar/gkx066 (2017).
- Ara A, Love RP, Follack TB, Ahmed KA, Adolph MB, Chelico L. Mechanism of Enhanced HIV Restriction by Virion Coencapsidated Cytidine Deaminases APOBEC3F and APOBEC3G. Journal of Virology, 91, pii: e02230-16 (2017).
- Wittkopp CJ, Adolph MB, Wu LI, Chelico L, Emerman M. A Single Nucleotide Polymorphism in Human APOBEC3C Enhances Restriction of Lentiviruses. PLoS Pathog 12(10): e1005865. (2016)
- Starrett, GJ, Luengas, EM, McCann, JL, Ebrahimi, D, Temiz, NA, Love, RP, Feng, Y, Adolph, MB, Chelico, L, Law, EK, Carpenter, MA, Harris, RS. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nature Communications. 7: 12918 (2016).
- Feng Y, Love RP, Ara A, Baig TT, Adolph MB, Chelico L. Natural polymorphisms and oligomerization of human APOBEC3H contribute to single-stranded DNA scanning ability. Journal of Biological Chemistry. 290: 27188-27203 (2015).
- Baig TT, Feng Y, Chelico L. Determinants of efficient degradation of APOBEC3 restriction factors by HIV-1 Vif. Journal of Virology. 88, 14380-14395 (2014).
- Feng Y, Baig TT, Love RP, Chelico L. Suppression of APOBEC3-mediated restriction of HIV-1 by Vif. Frontiers in Microbiology. Volume 5, Article 450, doi: 10.3389/fmicb.2014.0045 (2014).
- Ara A, Love RP, Chelico L. Different Mutagenic Potential of HIV-1 Restriction Factors APOBEC3G and APOBEC3F Is Determined by Distinct Single-Stranded DNA Scanning Mechanisms. PLoS Pathog 10(3): e1004024. doi:10.1371/journal.ppat.1004024 (2014)
- Adolph MB, Webb J, Chelico L. Retroviral Restriction Factor APOBEC3G Delays the Initiation of DNA Synthesis by HIV-1 Reverse Transcriptase. PLoSONE 8(5): e64196. doi:10.1371/journal.pone.0064196 (2013)
- Feng, Y, Love, R.P., Chelico, L. HIV-1 Viral infectivity factor (Vif) alters processive single-stranded DNA scanning of the retroviral restriction factor APOBEC3G. Journal of Biological Chemistry. 288: 6083-6094. (2013)
- Love R.P., Xu H., Chelico L. Biochemical Analysis of Hypermutation by the Deoxycytidine Deaminase APOBEC3A. Journal of Biological Chemistry. 287:30812-30822 (2012).
- Senavirathne G, Jaszczur M, Auerbach PA, Upton TG, Chelico L, Goodman MF, Rueda D. Single-stranded DNA Scanning and Deamination by APOBEC3G Cytidine Deaminase at Single Molecule Resolution. Journal of Biological Chemistry, 287:15826-15835. (2012)
- Feng Y. and Chelico L. Intensity of deoxycytidine deamination of HIV-1 proviral DNA by the retroviral restriction factor APOBEC3G is mediated by the non-catalytic domain. Journal of Biological Chemistry. 286, 11415-11426 (2011).
- Chelico, L., Prochnow, C., Erie, D.A., Chen, X.S., and Goodman, M.F. A structure-based model of APOBEC3G-catalyzed C deamination on single-stranded DNA. Journal of Biological Chemistry. 285, 16195-16205 (2010).
- Chelico, L., Pham, P., Petruska, J. and Goodman, M.F. Biochemical basis of immunological and retroviral responses to DNA-targeted cytosine deamination by AID and APOBEC3G. Journal of Biological Chemistry. 284, 27761-27765 (2009)
- Rausch, J., Chelico, L., Goodman, M.F. and Le Grice, S.F.J. Dissecting APOBEC3G substrate specificity by nucleoside analog interference. Journal of Biological Chemistry. 284, 7047-7058 (2009).
- Chelico, L., Pham, P. and Goodman, M.F. Stochastic properties of processive cytidine DNA deaminases AID and APOBEC3G. Philosophical Transactions of the Royal Society of London. B, Biological Sciences. 364, 583-593 (2009).
- Holden, L.*, Prochnow, C.*, Chang, Y.P.*, Bransteitter, R., Chelico, L., Sen, U., Stevens, R.C., Goodman, M.F. and Chen, X.S. The crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature. 456, 121-124 (2008). * These authors contributed equally.
- Chelico, L., Sacho, E.J., Erie, D.A. and Goodman, M.F. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. Journal of Biological Chemistry. 283, 13780-13791 (2008).
- Chelico, L., Pham, P., Calabrese, P. and Goodman, M.F. APOBEC3G DNA deaminase acts processively 3’ to 5’on single-stranded DNA. Nature Structural and Molecular Biology. 13, 392-399 (2006).